Quality check, reads filtering, trimming and deduplication was carried out with FastP 0.23.4Chen et al. (2018) and reports were merged with MultiQC 1.22.3. Reads processing parameters were optimized to maximal proportion of mapped reads against the reference HB27 assembly GCA_0000081251.
The statistics of the final reads, after QC-filtering and dedupication, are shown in the Table 1 below. The detailed FastP combined reports is available here.
Table 1. Statistics of fastp processed reads of HB27 TnSeq libraries
Library
File
Reads
% Duplication
Final reads (M)
Final reads (%)
GC %
Ppol_1
TNB-01_S1_L001_R1_001
2170192
78.20%
0.9
41.40%
59.40%
Ppol_2
TNB-09_S5_L001_R1_001
1820971
78.60%
1.7
92.10%
64.90%
HB27_1
TNB-03_S3_L001_R1_001
3196758
72.40%
2.7
83.90%
65.40%
HB27_2
TNB-07_S3_L001_R1_001
1515716
74.20%
1.3
88.30%
65.80%
The original sample looks slightly different, with less reads and length, as well as somewhat lower GC content. Note that HB27 chromosome has 1.894.877 bp and 69.5% GC and pTT27 has 232.605 and 69% GC.
Given that several samples were sequenced in the same batch, downplaying a technical problem with sequencing library o running, we hypothesize that some transposon insertions may be unstable in the context of Thermus.
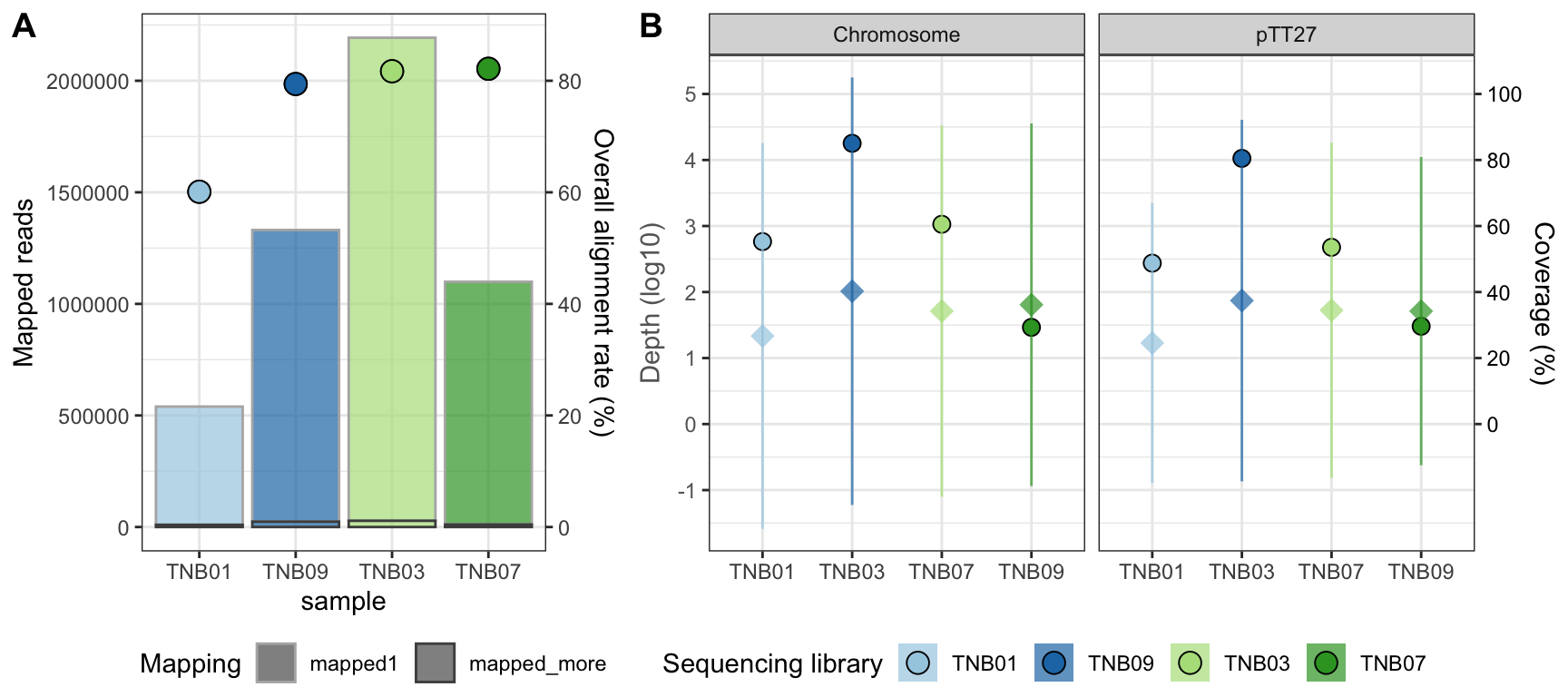
Figure 1. TnSeq libraries mapping statistics. (A) Reference sequence depth (diamonds, left axis) and coverage (round points, right axis). Samples corresponding to ppol and wt HB27 strains are colored in blues and greens, respectively. (B) Total number of mapped reads (bars, left axis) and overall alignment rate (points, right axis).
The mapping shows that overall 80% of the reads mapped once within the HB27 chromosome, giving rise to a good coverage of the HB27 genome, with roughly 100x coverage depth and 50% of coverage breadth. However, again, the sample TNB01 corresponding to ppol (Mother) is clearly different, with less reads and lower mapping rate.
Mapping coverage in HB27 genome
Let’s look in more detail the mapping per base pair, analyzed with Bedtools 2.30.0.
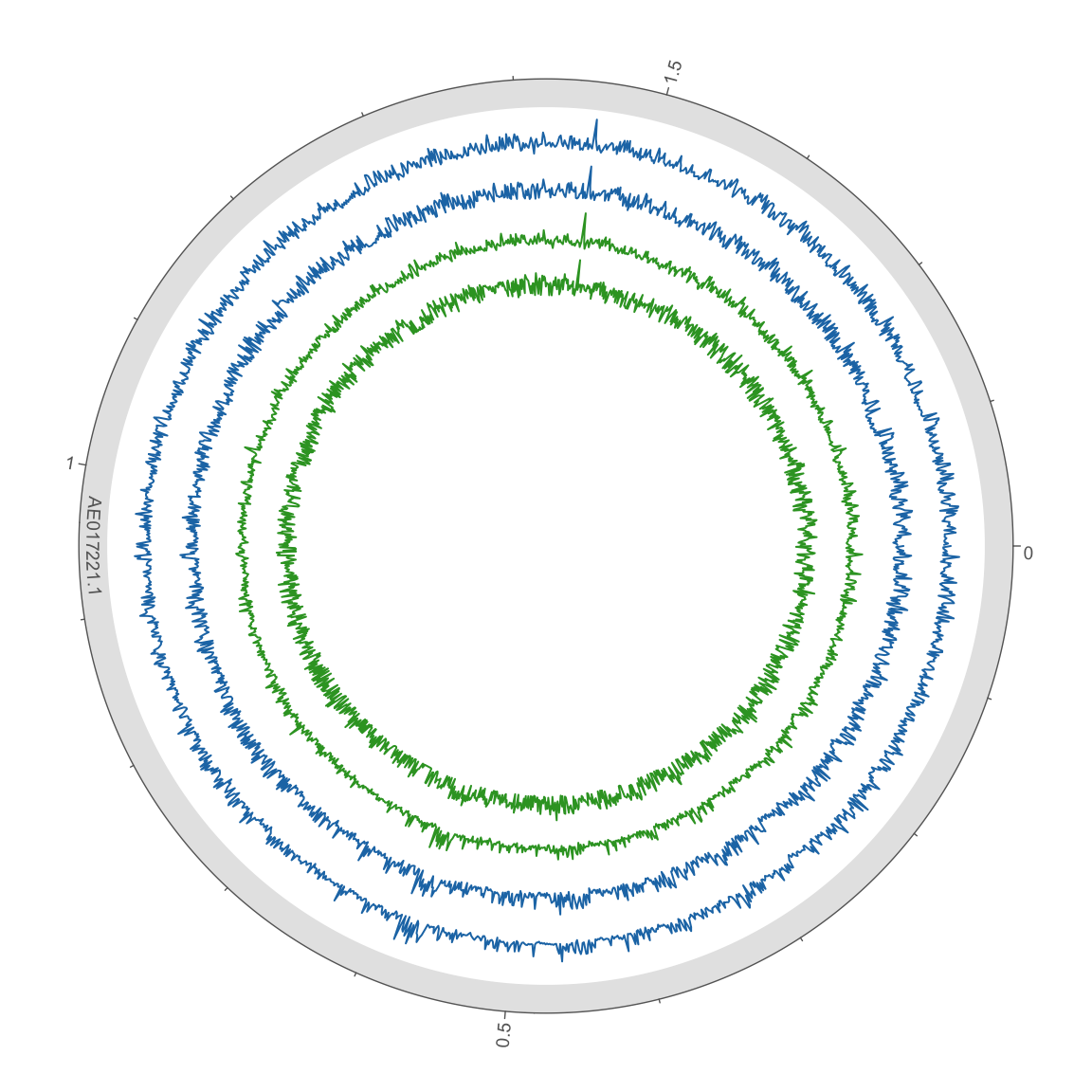
Figure 2. HB27 wt (green) and Ppol (blue) TnSeq libraries mapping in HB27 chromosome. Coverage is represented the mean cov per 1 kb widows in log10 scale. Outer circle scale in Mb.
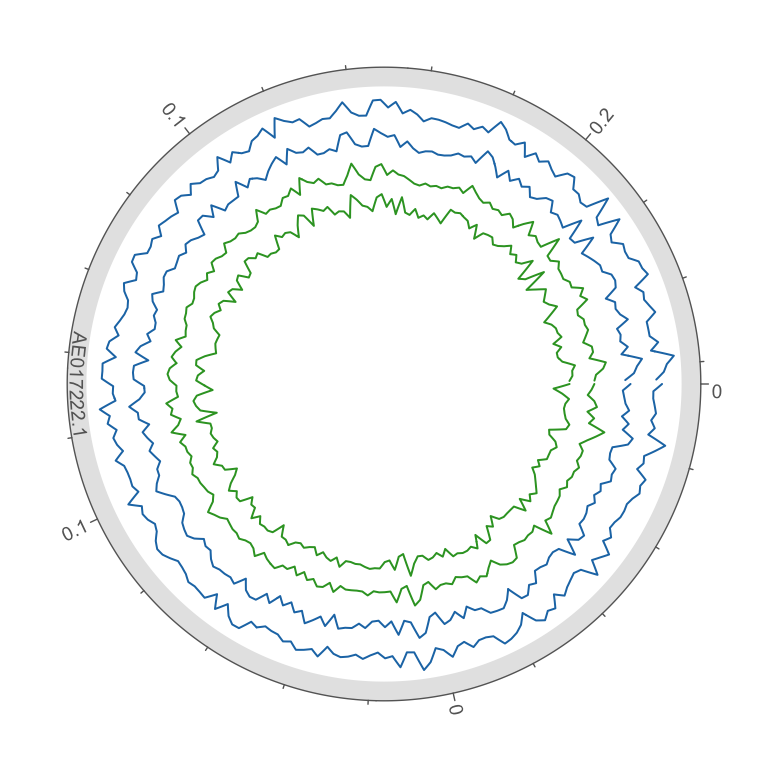
Figure 3. HB27 wt (green) and Ppol (blue) TnSeq libraries mapping in pTT27 plasmid. Coverage is represented the mean cov per 1 kb widows in log10 scale. Outer circle scale in Mb.
The above plots represent mean coverage per 1000 bp windows, full plots are in the folder bamdash and can be accessed with the following links, as interactive plots and SVG images.
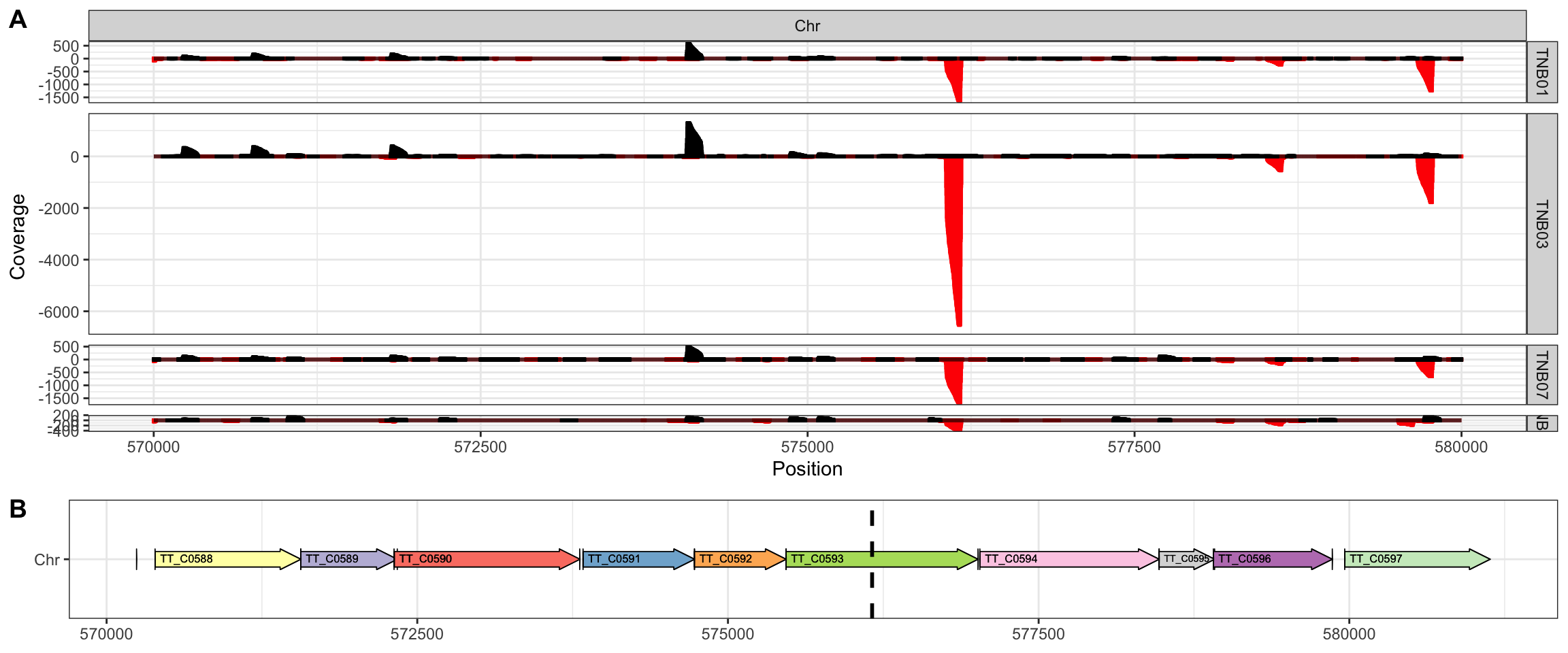
The two replicates have a smaller range than the primary samples (mother and daughter), but we can still recognize two regions with a very high frequency of insertions in the reverse strand, located at positions 576,160 and 1,457,335 of the chromosome and almost overlapping in the four samples.
We have now decided to investigate these regions in more detail. The first hotspot corresponds to gene TT_C0593 (Figure 3), which encodes a 5-carboxymethyl-2-hydroxymuconate semialdehyde dehydrogenase, an oxidoreductase involved in tyrosine metabolism that may be dispensable in rich media. This target region corresponds to a GC track, the preferred sequence for Tn5 (Green et al. 2012). However, as the coverage indicates multiple nearby integration sites, the high integration rate within TT_C0593 may indicate that this gene is not required rather than a transposase artifact.
Figure 4. TnSeq insertion hotspot (A) Detailed coverage (linear scale) in the region 570000-580000 of HB27 chromosome in each sample, Ppol (TNB01), Ppol_rep (TNB09), HB27 (TNB03) and HB27_rep (TNB07). (B) Annotated features in the detailed region. The vertical dashed line indicated the position of the highest coverage.
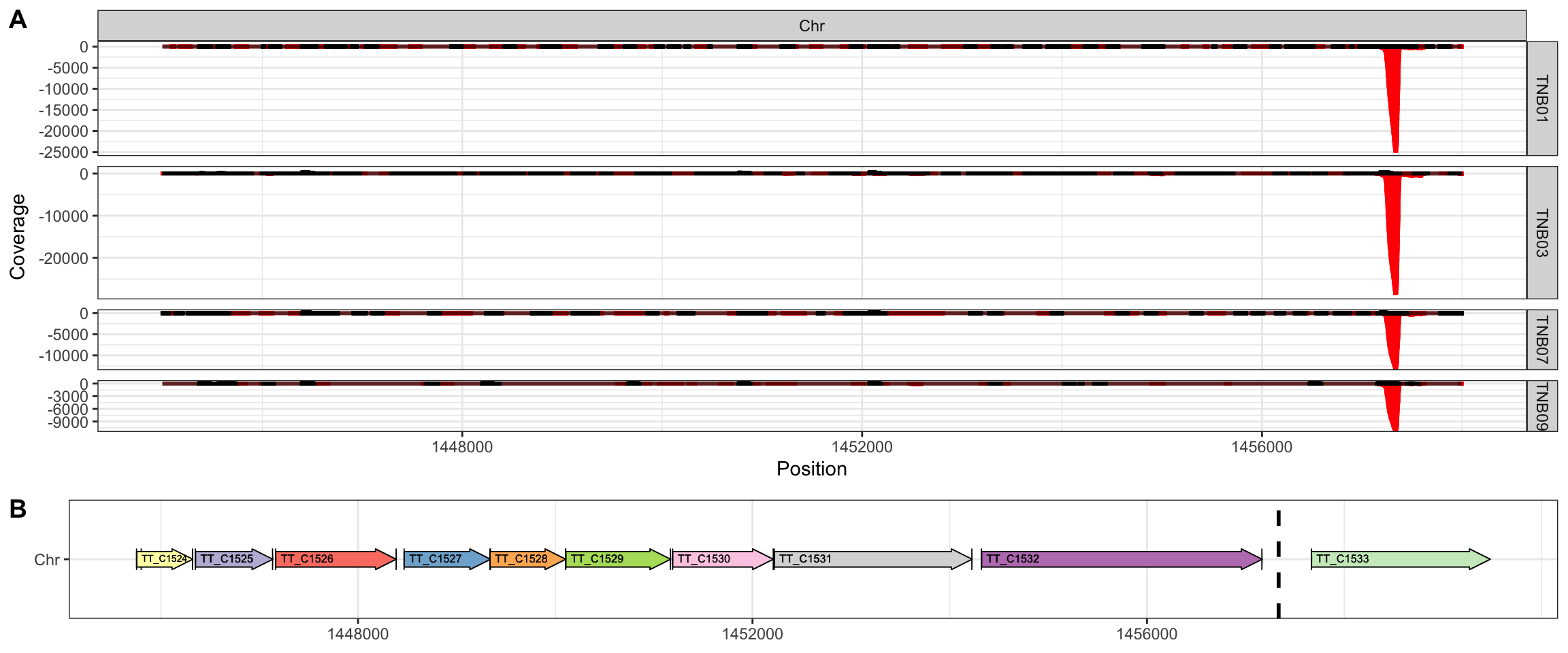
The second and more strong hotspot (Figure 5) is located in an intergenic region between locus TT_C1532 (glucosamine-fructose-6-phosphate aminotransferase) and TT_C1533 (S-layer protein). The sequence in this point appears to be more diverse but also contains GC traces. This hostpot is likely consequence of the use of S-layer protein promoter in front of the kanamycin resistance gene used for the transposition selection. Moreover, since we will normalize the counts per gene based on the mapped reads in the coding regions (see below), this promiscuous site will not affect our analysis.
Figure 5. TnSeq insertion hotspot (A) Detailed coverage in the region 1445000-1458000 of HB27 chromosome in each sequencing library: Ppol (TNB01), Ppol_rep (TNB09), HB27 (TNB03) and HB27_rep (TNB07). (B) Annotated features in the detailed region. The vertical dashed line indicated the position of the highest coverage.
Tn5 transposon insertions per gene and Z-score
Count integration events counts per gene and Z-score calculation
Now we are going to transform the mapped reads into hits per gene, to obtain a normalized insertion score, by the following steps:
We only consider insertions within the 10-90% interval of each gene, because insertions landing in the flanking sections of genes might give rise to truncated of chimeric proteins partially functional. The counts of Tn insertions will be obtained from the read mapping coordinates, considering the alignment start and the strand, after conversion of the BAM file to a tabulated format (BED).
We will normalize by the total number of mapped reads within the coding regions (80% central).
We will obtain a ratio of observed to expected Tn insertions.
We will make a log2-transformation in pseudocounts \(log_2(x+1)\) to avoid negative scores.
Finally, we will scale the data to favor sample comparison, using the R function scale() to obtain a final Z-score.
Thus, our score will be obtained with the following formula:
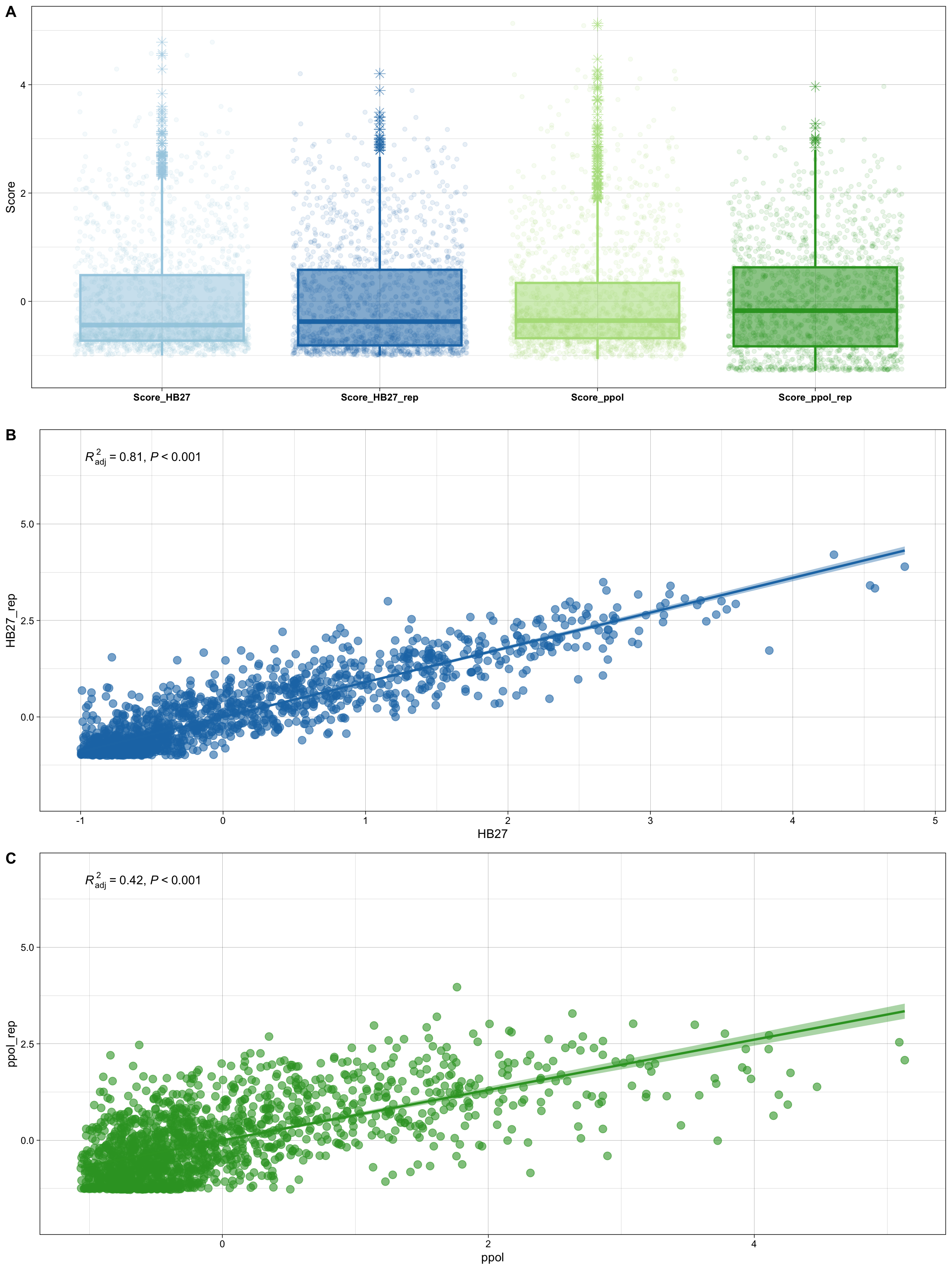
Figure 6. Tn insertion Z-scores per gene. (A) Boxplot of gene Z-scores per sample. (B & C) Correlation of Z-scores per gene between sample replicates.
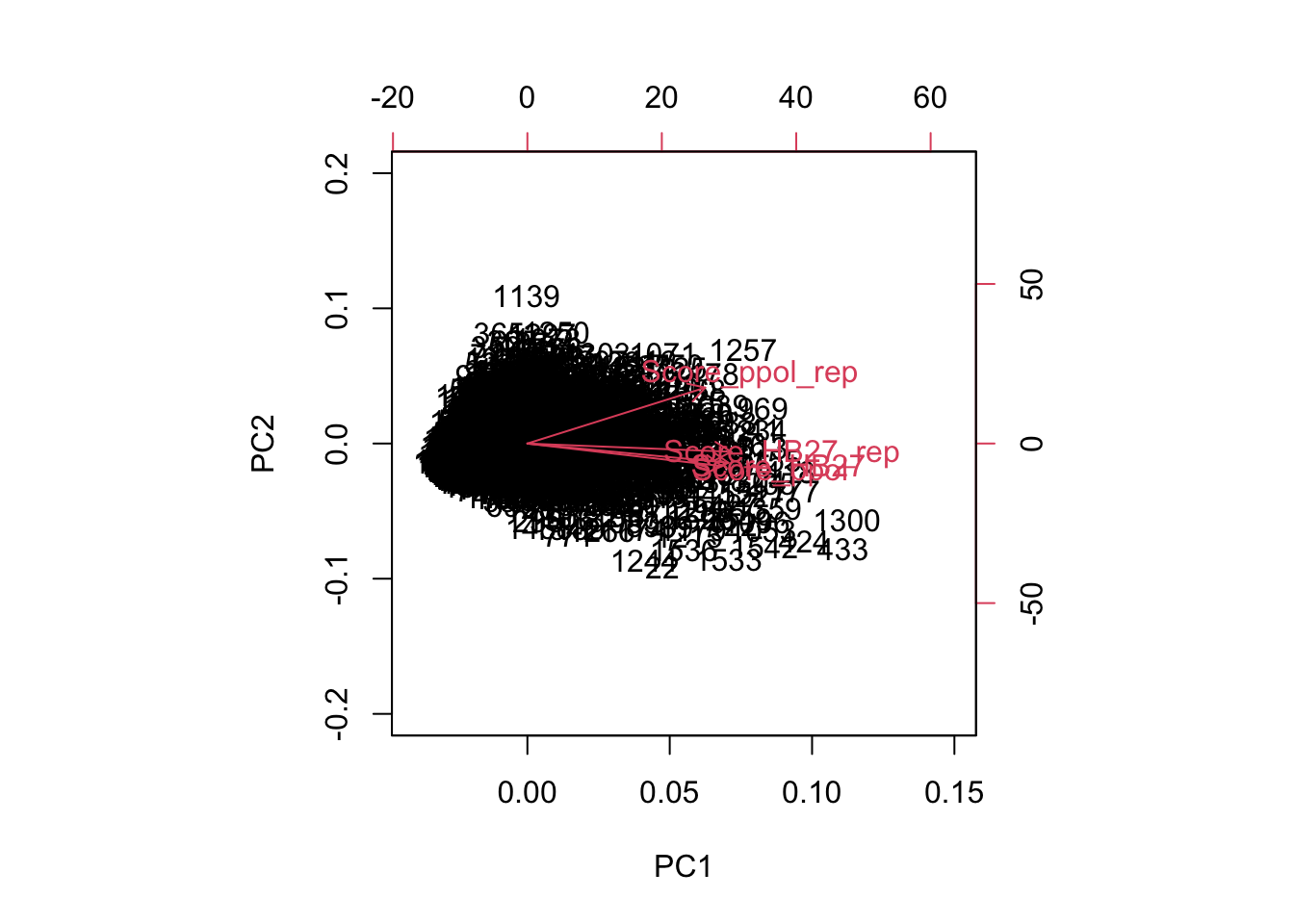
As we can see, the correlation between the two PPOL samples is only moderate, in line with the QC and mapping results. To analize this in more detail and compare the four samples, we used a PCA plot. As shown in Figure 7 below, the first variable is responsible for >80% of the data variability, with all samples being very similar in this direction. A second variable is responsible for ~10% of the data diversity, with half of this divergence attributable to sample Ppol_2 (TNB09). Interestingly, samples TNB01, TNB03 and TNB07 are quite similar, although sample TNB01 contains significantly fewer reads, suggesting that the sequencing library does not strongly influence our results.
Show code
tnseq.pca <-prcomp(na.omit(scores)[,1:4], center =TRUE, scale =TRUE) fviz_pca_biplot(tnseq.pca,col.var=rownames(tnseq.pca[[2]]),col.ind="grey90", label="var",labelsize=5,labelface="bold")+ylim(-4,4)+xlim(-2.5,9)+scale_color_brewer(palette="Paired")+theme_pubclean()+theme(legend.position="none")
Figure 7. Samples PCA plot. Note that only complete cases (genes with at least one Tn5 insertion in all samples) can be used.
In order to analyze in detail the difference between samples, we construct also a plot in which we plot the Z-scores for all genes in all samples.
Figure 8. Comparison of Tn insertion Z-scores per gene. Values were sorted by the average of all samples. This is an interactive plot: Put your mouse pointer over any point and you will see a pop-up label with the Z-score and the Gene info.
It is striking that we do not see a step that marks the boundary between the group of non-essential genes, which are expected to have a higher score, and the essential genes with very low or no Tn5 insertions and thus a low score.
Summary stats
In the following table you can find a summary of the main raw insertion stats. The average number of insertions within the 80% central portion of the gene was around 1.8x10^5, with roughly 4.7 x10^4 unique insertion sites, that is an average of 1 unique insertion site per 44.8 bp.
Show code
kk <-cbind(insertion_table[[1]], "HB27_1")kk <-rbind(kk,cbind(insertion_table[[2]], "HB27_2"))kk <-rbind(kk,cbind(insertion_table[[3]], "Ppol_1"))kk <-rbind(kk,cbind(insertion_table[[4]], "Ppol_2"))#unique insertions per sample and per strandunique_insertions <-aggregate(data=kk, i.start ~ Strand+V2, function(x) length(unique(x)))#unique insertions total per sampleunique_insertions <-aggregate(unique_insertions,unique_insertions$i.start~unique_insertions$V2,sum)#allresumen <-cbind(fastp[,c(1,4)],as.vector(table(kk$V2)),unique_insertions[,2])#normalized & ratioresumen$normalized <- resumen[,4]/resumen[,2]resumen[,2] <- resumen[,2] *1000000resumen$ratio <-paste(as.character(round(resumen[,4]*100/resumen[,3],1)),"%")resumen[5,] <-c("Mean",apply(resumen[,2:5],2,mean),paste(as.character(round(mean(resumen[,4]*100/resumen[,3]),1)),"%"))resumen[,2] <-as.numeric(resumen[,2])resumen[,3] <-as.numeric(resumen[,3])resumen[,4] <-as.numeric(resumen[,4])resumen[,5] <-as.numeric(resumen[,5])as.data.frame(format(resumen[c(1,4,3,2,5),c(1:4,6,5)], scientific =TRUE, digits =3)) %>%kbl(align ="c", row.names =FALSE,col.names=c("Sample","Filtered Reads","Insertions 80% ORF","Unique insertions 80% ORF","Ratio unique","Normalized Unique insertions per M Reads"), caption ="Table 3. Summary of TnSeq statistics") %>%kable_styling(bootstrap_options ="striped", full_width = F, position ="center") %>%column_spec(1, bold = T, italic=T, color=c(my.cols,"grey30"))
Table 3. Summary of TnSeq statistics
Sample
Filtered Reads
Insertions 80% ORF
Unique insertions 80% ORF
Ratio unique
Normalized Unique insertions per M Reads
Ppol_1
9.00e+05
1.05e+05
3.69e+04
35.2 %
4.10e+04
Ppol_2
1.70e+06
1.64e+05
4.39e+04
26.7 %
2.58e+04
HB27_2
1.30e+06
2.88e+05
7.14e+04
24.8 %
5.49e+04
HB27_1
2.70e+06
1.56e+05
3.77e+04
24.1 %
1.40e+04
Mean
1.65e+06
1.78e+05
4.75e+04
27.7 %
3.39e+04
Insertions in key genes
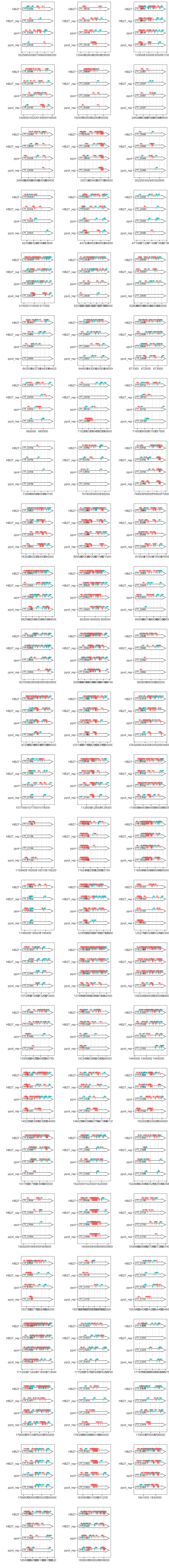
We now decided to check some genes, expected to be essential, and see their score and the number and location of the insertions in each sample.
Figure 9. Tn insertions in key HB27 genes in forward (blue) and reverse (pink) strands. Only insertions between 10-90% central gene interval considered for Z-score calculation are displayed.
Green, Brian, Christiane Bouchier, Cécile Fairhead, Nancy L. Craig, and Brendan P. Cormack. 2012. “Insertion Site Preference of Mu, Tn5, and Tn7 Transposons.”Mobile DNA 3 (1): 3. https://doi.org/10.1186/1759-8753-3-3.
Footnotes
For Tn insertions purposes, we merged the TT_C0145 and TT_C0146 genes, based on (unpublished) evidence from Mencía lab.↩︎